Prelude
When people first enter genomics, sequencing can look like a menu of technologies: whole genome sequencing (WGS), whole exome sequencing (WES), RNA-seq, targeted panels, single-cell, methylation sequencing, and more.
It’s tempting to treat these as interchangeable tools with different price points. But that misses the central idea:
Sequencing modalities exist because biological questions are different. And different questions require different tradeoffs in resolution, scale, sensitivity, cost, turnaround time, and interpretability.
In other words, the most important question is not “Which sequencing method is best?” It’s “Best for what, and why?”
This post focuses on sequencing-based genomic and transcriptomic modalities; it does not cover proteomics, metabolomics, or other non-sequencing omics.
⚡ For the Impatient
Start with your biological question. Click to reveal which modality fits and why.
1 I need to discover unknown or non-coding variants
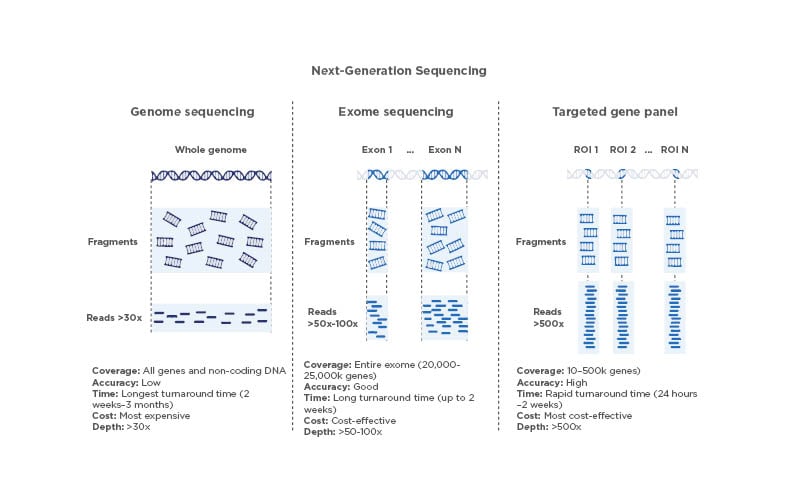
WGS Broadest hypothesis-free view. Captures structural variants, regulatory regions, and non-coding biology that exome and panels miss.
Tradeoff: Higher cost, storage, and interpretation burden for that breadth.2 I need broad coding-region coverage at reasonable cost
WES Targets ~1–1.5% of the genome (protein-coding exons). Cost-efficient for Mendelian disease and broad gene screening without full-genome complexity.
Tradeoff: Misses non-coding, structural, and capture-challenging regions.3 I need high sensitivity in known genes or hotspots
Targeted Panel Concentrates sequencing depth on predefined loci. Maximizes sensitivity for low-VAF variants, ctDNA, and clinically actionable targets.
Tradeoff: Blind to novel genes or unexpected biology outside the panel.4 I need to know what the cell is actually doing
RNA-seq Profiles transcript abundance, splicing, fusions, and pathway activity. Reveals functional state that DNA sequence alone cannot explain.
Tradeoff: RNA is context-dependent and unstable; transcript levels ≠ protein levels.5 I need to know which cells are driving the signal
Single-cell Resolves cell-level heterogeneity — rare clones, transitional states, and subpopulations that bulk averaging erases.
Tradeoff: Higher noise (ambient RNA, doublets), cost, and loss of spatial context.6 I need to know why a gene is on or off
Epigenomic Measures regulatory layers — methylation, chromatin accessibility, histone marks — that control gene activity beyond DNA sequence.
Tradeoff: Highly context-specific; harder to translate into direct clinical action.7 I need to monitor disease non-invasively over time
Liquid Biopsy Sequences ctDNA/cfDNA from blood. Enables serial monitoring of treatment response, resistance, and minimal residual disease without tissue biopsy.
Tradeoff: Low tumor fraction limits sensitivity; shedding varies by tumor type and stage.8 I need to know where in the tissue the biology is happening
Spatial Transcriptomics Maps gene expression to tissue architecture — immune exclusion zones, invasion fronts, regional programs — preserving the spatial context dissociation destroys.
Tradeoff: Resolution varies by platform; cost and tissue handling are demanding.9 I need a fast, deep readout of a small known region
Amplicon PCR-amplifies specific targets for deep, efficient sequencing. Ideal for 16S/ITS profiling, hotspot testing, and pathogen surveillance at scale.
Tradeoff: Strictly limited to predefined regions; PCR bias can skew representation.10 I need to know what organisms are in a mixed sample
Metagenomics Profiles community composition, strain variation, and functional potential directly from environmental or clinical samples without culture.
Tradeoff: Host contamination, reference-database dependence, and presence ≠ activity.11 I need to profile immune clonality and receptor diversity
TCR/BCR-seq Sequences rearranged immune receptors to reveal clonal expansion, repertoire breadth, V(D)J usage, and antigen-driven selection.
Tradeoff: Focused on receptor biology; linking clonotypes to antigen specificity requires further evidence.12 One molecular layer isn't enough to explain this biology
Multiome / Multimodal Jointly profiles RNA + chromatin, RNA + protein, or other combinations from the same cell or tissue section. Connects expression to regulation to identity.
Tradeoff: Shallower depth per modality; sometimes separate optimized assays + computational integration outperforms joint profiling.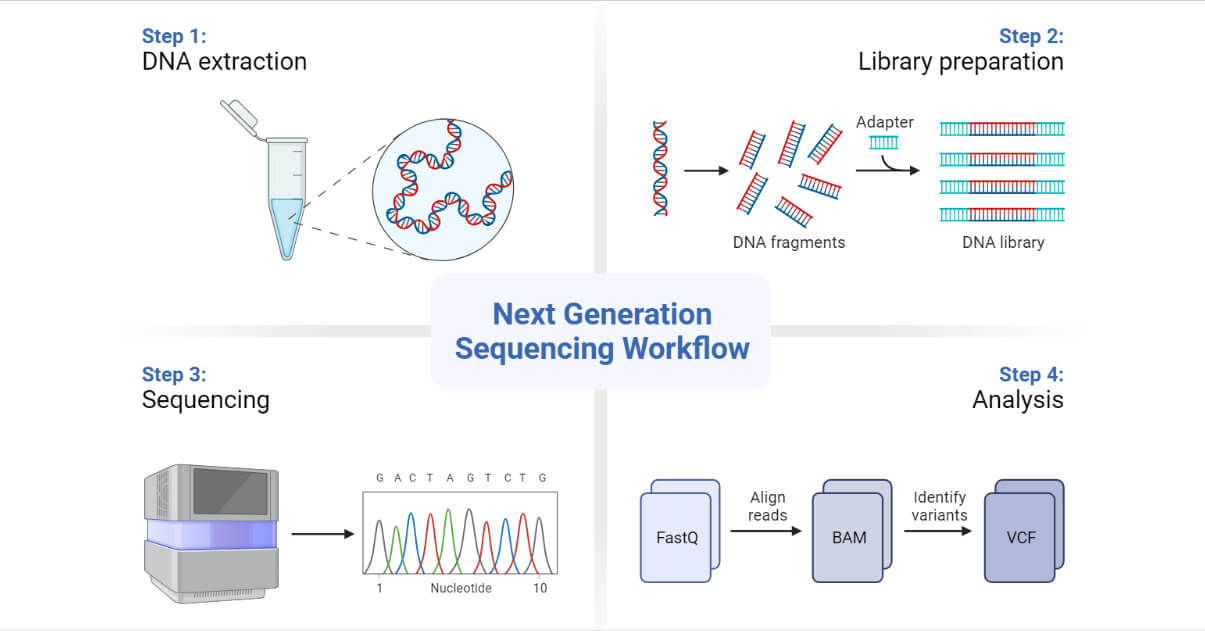
Sequencing modality is a question-design problem
Every sequencing experiment is a measurement strategy. Before choosing a modality, you’re implicitly deciding:
- What molecule matters most? (DNA, RNA, chromatin, methylation marks)
- What variation matters most? (SNVs, indels, CNVs, structural variants, fusions, expression changes)
- At what scale? (single gene, panel, exome, whole genome, single cell, tissue-level)
- At what sensitivity? (rare variant detection, low VAF, minimal residual disease)
- Under what constraints? (cost, sample quality, turnaround time, computational burden)
That’s the “why” behind sequencing modalities: each one is optimized around a particular biological or clinical decision.
1) Whole Genome Sequencing (WGS): Why sequence everything?
What it captures
WGS interrogates essentially all genomic DNA—coding and non-coding regions, mitochondrial DNA (depending on prep/analysis), and broad structural context.
Why choose it
You choose WGS when the question is:
- “What are we missing if we only look where we expect to find something?”
- “Could the answer be outside the exome?”
- “Do we need a unified view of variant classes?”
WGS is especially powerful when you care about:
- Structural variants
- Copy number changes
- Non-coding/regulatory variants
- Complex rearrangements
- Repeat regions (with limitations in short-read WGS)
- Genome-wide context instead of targeted snapshots
The deeper “why”
Biology is not confined to exons. Many disease mechanisms arise from:
- regulatory elements,
- intronic splice effects,
- enhancers,
- promoters,
- non-coding RNAs,
- and structural rearrangements that disrupt genomic architecture.
WGS exists because hypothesis-limited sequencing can fail when biology is unexpected.
Tradeoffs
- More data, higher storage/compute burden
- Higher cost than targeted approaches
- Interpretation can be harder (many variants of uncertain significance)
- Coverage depth may be lower than targeted tests at the same budget
Bottom line: WGS is a discovery-forward modality. You pay for breadth because you don’t want biology pre-filtered.

2) Whole Exome Sequencing (WES): Why focus on coding regions?
What it captures
WES enriches and sequences primarily protein-coding exons (~1–2% of the genome, depending on capture design).
Why choose it
You choose WES when the question is:
- “Can we capture most high-impact Mendelian variants efficiently?”
- “We need broad gene coverage, but not full-genome cost/complexity.”
WES became popular because a large proportion of currently characterized disease-causing variants in rare disorders lie in coding regions and directly alter protein sequence or canonical splicing.
The deeper “why”
WES is a cost-efficient compromise between targeted panels and WGS:
- broader than panels (less biased by current knowledge),
- cheaper and easier to interpret than WGS,
- often sufficient for many rare disease workflows.
It reflects an important truth in genomics operations: sometimes the best assay is not the most comprehensive one, but the one that is comprehensive enough to answer the question reliably and at scale.
Tradeoffs
- Uneven capture efficiency
- Misses many non-coding variants
- Poorer performance in GC-rich, repetitive, or capture-challenging regions
- Structural variant detection is limited vs WGS
- Exome definitions vary by kit/version
Bottom line: WES exists because many clinical and research questions need breadth across genes, but not the full cost and interpretive burden of the entire genome.

3) Targeted Gene Panels: Why not sequence everything?
What it captures
A defined set of genes/regions relevant to a disease area (e.g., oncology hotspots, cardiomyopathy genes, inherited cancer genes).
Why choose it
You choose a panel when the question is:
- “We need a fast, focused, high-confidence answer in known loci.”
- “Clinical actionability matters more than discovery.”
- “We need very deep coverage for low-frequency variants.”
Panels are ideal when:
- the disease biology is relatively well characterized,
- turnaround time matters,
- sample input is limited,
- and you need analytical sensitivity (e.g., low VAF in tumor samples or ctDNA).
The deeper “why”
Panels exist because depth beats breadth when the signal is rare.
If you suspect variants in a known set of genes and need to detect mutations at low allele fractions, spending reads across the entire genome is inefficient. Panels concentrate sequencing power exactly where it matters.
This is why panels remain foundational in many clinical labs despite the rise of WGS/WES.
Tradeoffs
- Can miss novel genes or atypical biology
- Must be updated as knowledge evolves
- Design biases can influence performance
- Limited utility if phenotype is unclear or heterogeneous
Bottom line: Panels are decision-optimized assays. They exist to maximize sensitivity, speed, and actionability in known biology.
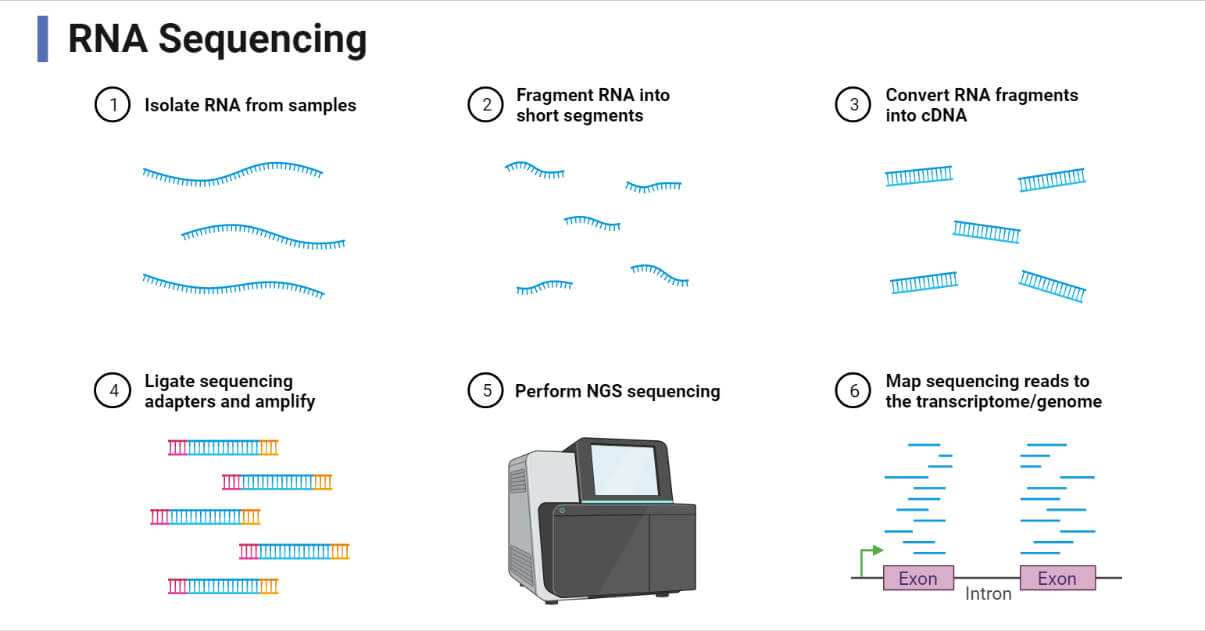
4) RNA Sequencing (RNA-seq): Why sequence transcripts instead of DNA?
What it captures
RNA-seq profiles transcript abundance and can reveal:
- gene expression changes,
- alternative splicing,
- fusion transcripts,
- allele-specific expression,
- transcript isoforms (especially with long-read RNA approaches).
Why choose it
You choose RNA-seq when the question is:
- “What is the cell actually doing?”
- “Is this variant functionally affecting expression or splicing?”
- “What pathways are active?”
DNA tells you what could happen. RNA tells you what is being executed (with caveats).
The deeper “why”
Many phenotypes are driven less by sequence changes alone and more by gene regulation and cell state. Two samples can have similar DNA but very different biology because of transcriptional programs.
RNA-seq exists because genotype is not the same as phenotype.
It is also invaluable for:
- validating the impact of suspected splice variants,
- identifying expressed fusions in cancer,
- understanding treatment response,
- and characterizing disease mechanisms beyond static DNA variation.
Tradeoffs
- RNA is less stable than DNA (sample handling matters)
- Expression is context-dependent (cell type, timing, environment)
- Tissue availability can be limiting
- Transcript abundance ≠ protein abundance
- Batch effects can be substantial
Bottom line: RNA-seq exists because DNA sequence alone often cannot explain biological behavior.
5) Single-Cell Sequencing: Why average signals can mislead
What it captures
Single-cell modalities (scRNA-seq, scATAC-seq, multiome, etc.) profile individual cells rather than bulk tissue averages.
Why choose it
You choose single-cell methods when the question is:
- “Which cells are driving this signal?”
- “Is this sample heterogeneous?”
- “Are rare populations or transitional states present?”
The deeper “why”
Bulk sequencing produces an average. But biology—especially tumors, immune responses, development, and tissue regeneration—is often governed by heterogeneity.
An average can hide:
- rare resistant tumor clones,
- activated immune subsets,
- developmental intermediates,
- or pathological cell states.
Single-cell sequencing exists because cell identity and state matter, and averages can erase them.
Tradeoffs
- Higher technical noise/dropout
- More complex analysis and interpretation
- More expensive per informative sample than bulk in many settings
- Dissociation bias and loss of spatial context (unless paired with spatial methods)
Bottom line: Single-cell methods are chosen when “who is doing what” matters more than the average signal.

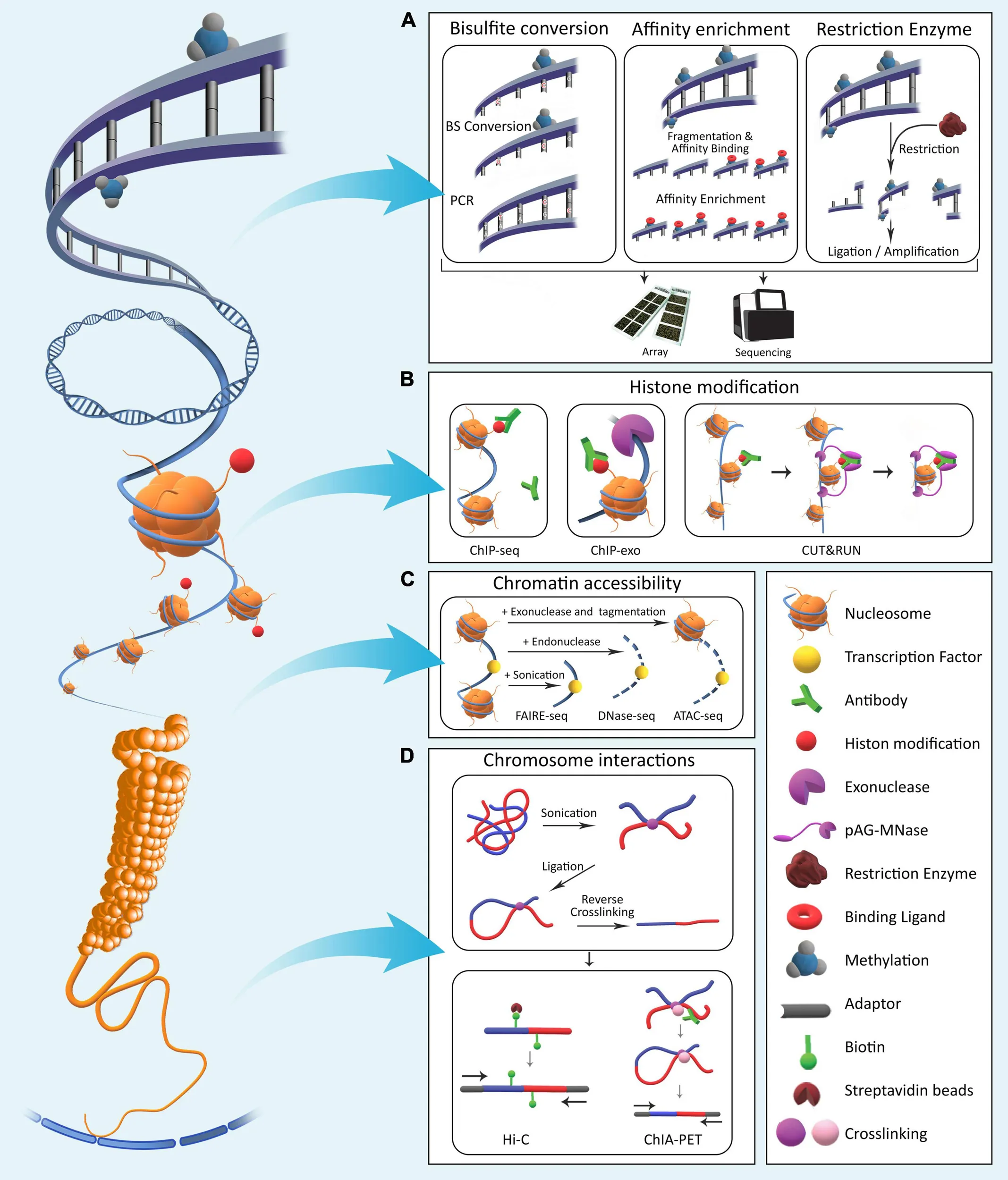
6) Epigenomic Sequencing (e.g., methylation, chromatin accessibility): Why sequence beyond sequence?
What it captures
These modalities measure regulatory layers such as:
- DNA methylation patterns,
- chromatin accessibility (ATAC-seq),
- histone modifications (ChIP-seq-like approaches),
- 3D genome interactions (Hi-C and related methods).
Why choose it
You choose epigenomic sequencing when the question is:
- “Why is this gene on or off?”
- “What regulatory state defines this cell or disease?”
- “Is gene regulation altered without a DNA sequence change?”
The deeper “why”
Sequence is the blueprint. Epigenetics and chromatin are part of the control system.
Many diseases, especially cancer and neurodevelopmental disorders, involve dysregulation that is not explained by DNA mutations alone. Epigenomic assays exist because biological function depends on genomic context and accessibility, not just nucleotide order.
Tradeoffs
- Strong dependence on sample quality and cell composition
- Interpretation can be highly context-specific
- Regulatory maps can be harder to translate into direct clinical action
Bottom line: These modalities exist because biology is regulated, not just encoded.
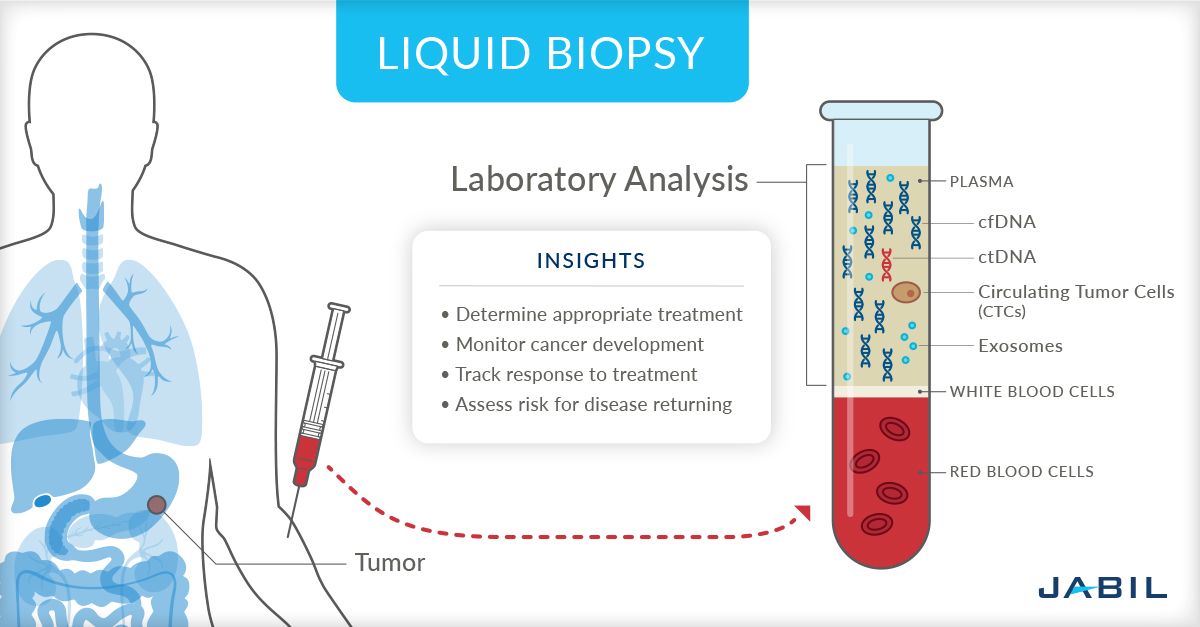
7) Liquid Biopsy Sequencing (ctDNA): Why sequence what’s circulating?
What it captures
Circulating tumor DNA (ctDNA) or cell-free DNA (cfDNA) from blood, often using targeted ultra-deep sequencing (sometimes methylation/signature-based approaches).
Why choose it
You choose liquid biopsy when the question is:
- “Can we monitor disease non-invasively?”
- “Can we detect residual disease or relapse earlier?”
- “Is tissue biopsy risky, unavailable, or unrepresentative?”
The deeper “why”
Tumors evolve, and tissue biopsies are invasive snapshots from one place and one time. ctDNA sequencing exists because disease monitoring needs repeatable, low-burden sampling.
It’s especially valuable for:
- treatment response monitoring,
- resistance mutation tracking,
- minimal residual disease (MRD),
- and sometimes early detection workflows (still highly context-dependent).
Tradeoffs
- Low tumor fraction can limit sensitivity
- Requires careful error suppression and assay design
- Biology of shedding varies by tumor type and stage
- Negative result may not exclude disease
Bottom line: Liquid biopsy modalities exist because longitudinal monitoring is a different problem than one-time diagnosis.
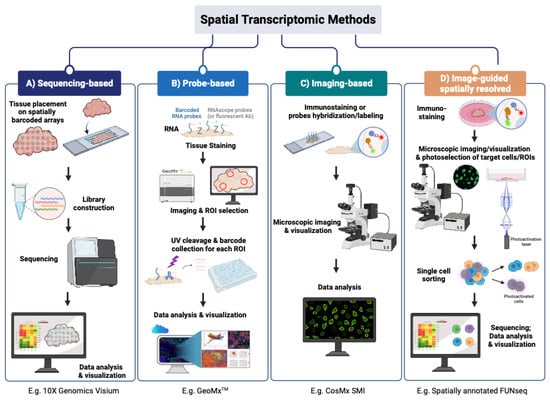
8) Spatial Transcriptomics: Why preserve location instead of dissociating cells?
What it captures
Spatial transcriptomics measures gene expression while preserving where signals occur within a tissue section. Depending on the platform, it can provide:
- gene expression mapped back to tissue architecture,
- regional transcriptional programs,
- cell neighborhood information,
- tumor–stroma or immune–epithelial interactions,
- and context for where specific cell states emerge.
Why choose it
You choose spatial methods when the question is:
- “Where in the tissue is this biology happening?”
- “Are these cell states interacting in a meaningful local context?”
- “Does tissue organization matter for interpreting this signal?”
The deeper “why”
Single-cell sequencing solved one major problem in biology: averages hide heterogeneity. But it created another: dissociation removes spatial context.
A dissociated cell can tell you what it is doing, but not where it came from, what it was touching, or what local microenvironment shaped its behavior.
Spatial transcriptomics exists because location is often part of the biological mechanism.
This matters especially in:
- tumors, where immune exclusion or invasion fronts are spatial phenomena,
- fibrosis, where remodeling is often region-specific,
- development, where gradients and boundaries define cell fate,
- and tissue injury/repair, where microenvironment determines response.
Tradeoffs
- Resolution varies by platform (some measure spots or regions, not true single cells)
- Analysis is more complex than standard bulk RNA-seq
- Cost can be substantial
- Tissue handling, section quality, and histology integration matter a lot
Bottom line: Spatial transcriptomics exists because biology is organized in space, and removing that context can remove the mechanism.
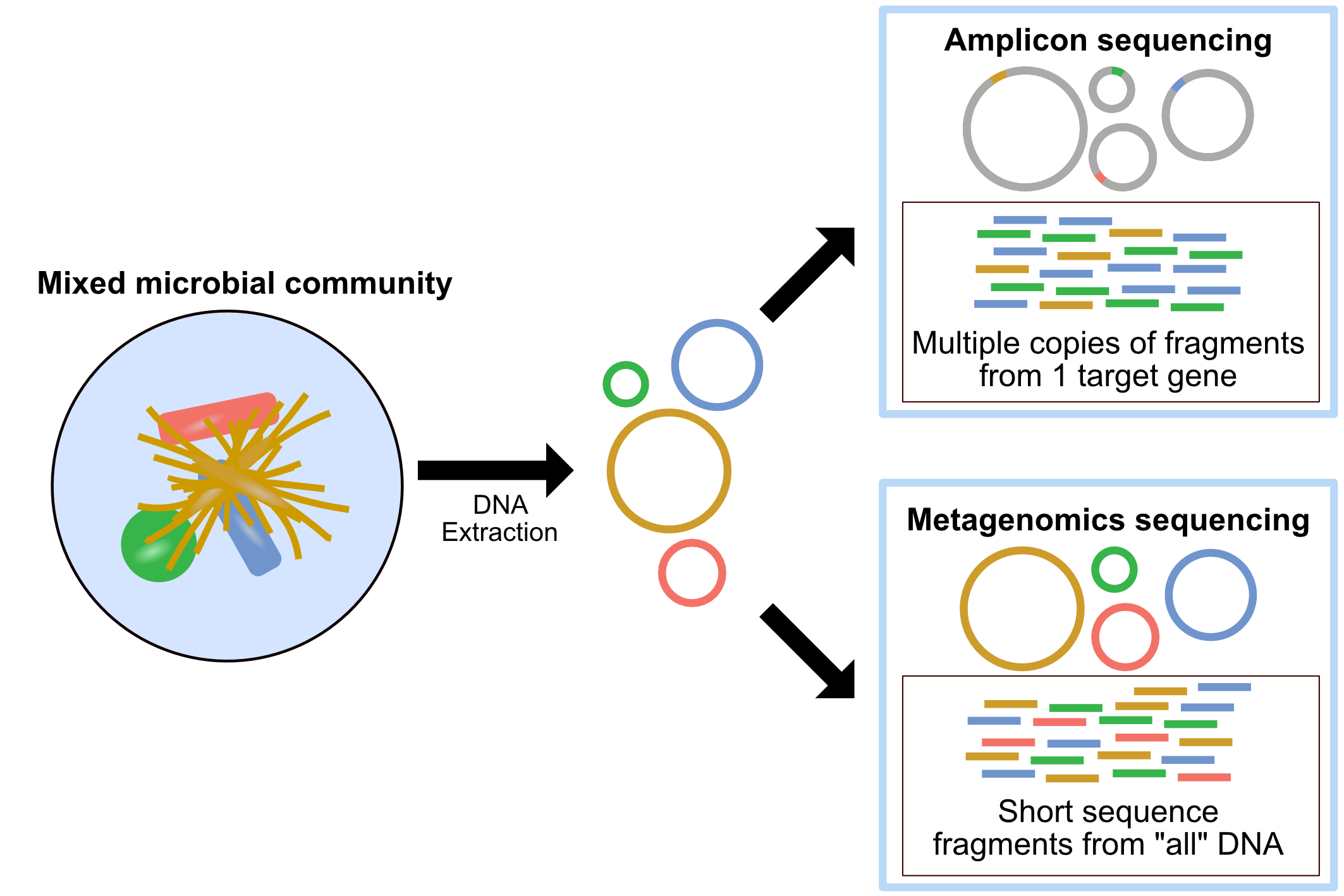
9) Amplicon Sequencing: Why amplify specific regions instead of sequencing broadly?
Why amplify specific regions instead of sequencing broadly?
What it captures
Amplicon sequencing targets and sequences specific predefined regions using PCR amplification before sequencing. Depending on the design, this can include:
- small genomic loci,
- mutation hotspots,
- microbial marker regions (e.g., 16S rRNA or ITS),
- viral genomic segments,
- or focused panels of short regions relevant to a specific assay.
Why choose it
You choose amplicon sequencing when the question is:
- “Do we need a fast, focused readout of a known region?”
- “Can we answer this question by interrogating a small number of targets very deeply?”
- “Is scale, simplicity, or sensitivity more important than breadth?”
Amplicon sequencing is especially useful when the target regions are already known, many samples need to be processed efficiently, input material is limited, or deep coverage is needed over a small region.
The deeper “why”
Not every sequencing question requires broad discovery. Sometimes the real need is a highly efficient measurement of a small, well-defined target.
Amplicon sequencing exists because some questions are best answered by concentrating sequencing effort on exactly the region of interest.
This is why it is widely used for:
- microbiome marker profiling (e.g., 16S or ITS),
- hotspot mutation testing,
- targeted pathogen surveillance,
- and focused validation of known loci.
In these settings, sequencing the whole genome, exome, or transcriptome would add cost and complexity without necessarily improving the decision.
Tradeoffs
- Strongly limited to predefined regions
- PCR bias can affect representation
- Poor fit for novel discovery outside target loci
- Primer design and amplicon dropout can influence results
- Often less informative about broader genomic context
Bottom line: Amplicon sequencing exists because some questions are narrow, known in advance, and best answered with focused depth rather than broad coverage.
10) Metagenomic Sequencing: Why sequence mixtures of organisms instead of one genome?
What it captures
Metagenomic sequencing profiles genetic material from mixed microbial communities directly from a sample, without requiring culture. Depending on the approach, it can reveal:
- which organisms are present,
- their relative abundance,
- strain-level variation,
- functional genes and pathways,
- and sometimes antimicrobial resistance or virulence features.
Why choose it
You choose metagenomic sequencing when the question is:
- “What organisms are in this sample?”
- “What is this microbial community capable of doing?”
- “Are we missing organisms that culture-based methods fail to detect?”
The deeper “why”
Many biological and clinical samples are not one genome—they are ecosystems. In those settings, the question is not just what mutation a single organism carries, but which organisms coexist, compete, cooperate, and contribute to phenotype.
Metagenomic sequencing exists because community composition and collective function can be the biology.
This is especially useful for:
- microbiome studies,
- environmental samples,
- infectious disease cases with unclear etiology,
- and settings where culture is slow, biased, or incomplete.
Tradeoffs
- Host contamination can reduce sensitivity
- Interpretation can be difficult in low-biomass samples
- Taxonomic assignment depends on reference databases and analysis choices
- Presence does not always equal activity or causation
Bottom line: Metagenomic sequencing exists because some biological questions are about communities, not individual genomes.
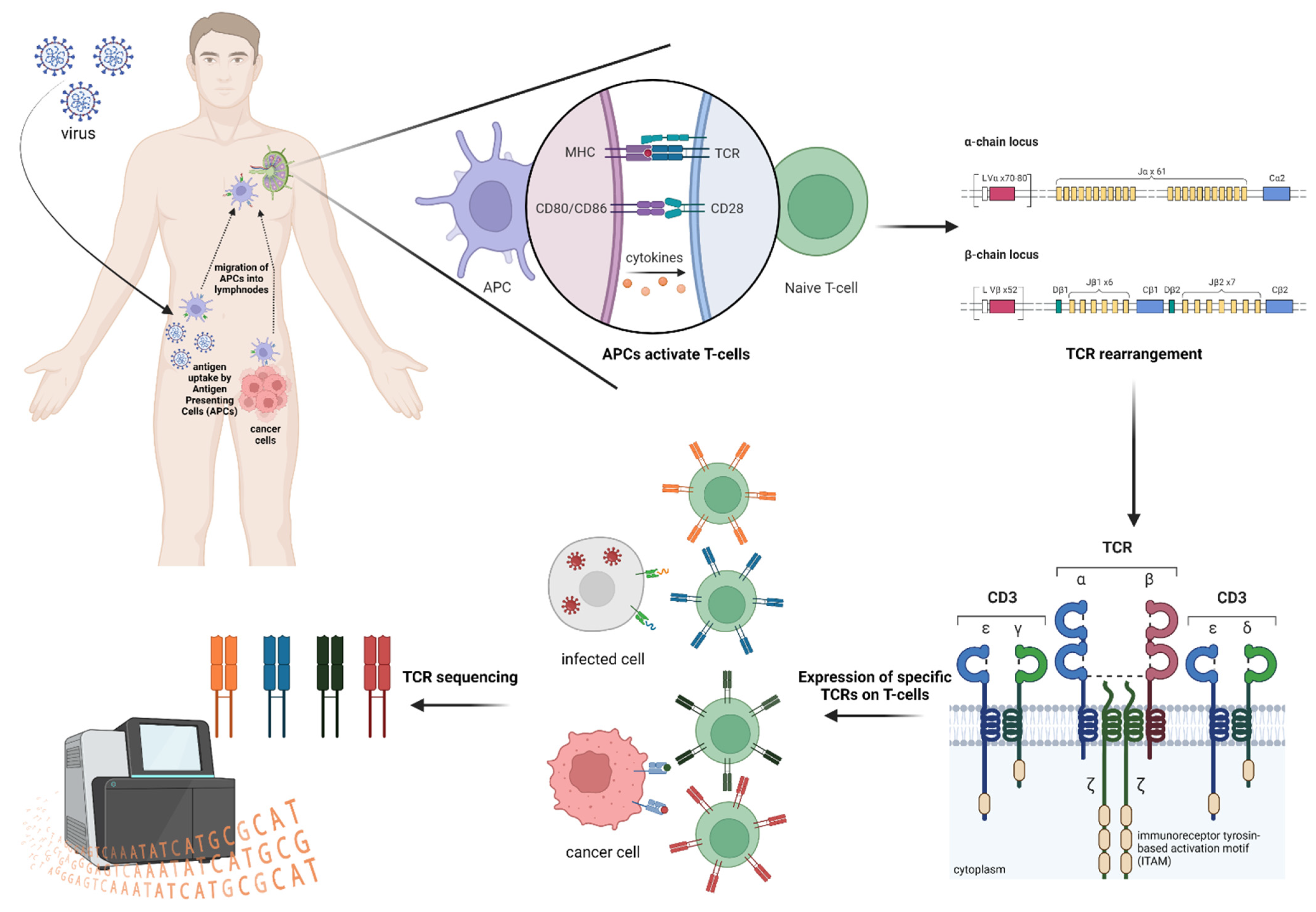
11) Immune Repertoire Sequencing (TCR/BCR-seq): Why sequence receptor diversity specifically?
What it captures
Immune repertoire sequencing profiles rearranged T-cell receptor (TCR) or B-cell receptor (BCR / immunoglobulin) sequences, especially the highly variable regions that define clonotypes. Depending on the assay, it can reveal:
- clonal diversity,
- clonal expansion,
- dominant receptor sequences,
- V(D)J usage,
- somatic hypermutation patterns (especially in B cells),
- and lineage relationships across immune populations.
Why choose it
You choose TCR/BCR sequencing when the question is:
- “Which immune clones are expanding?”
- “Is the immune response broad, restricted, or antigen-driven?”
- “Are specific lymphocyte clones associated with disease, treatment response, or persistence?”
This is especially valuable when studying immune responses to infection, cancer immunology, autoimmune disease, vaccine responses, minimal residual disease in lymphoid malignancies, and immune reconstitution over time.
The deeper “why”
The adaptive immune system works through receptor diversity and clonal selection. In many cases, the most important biological signal is not average gene expression, but which receptor-defined clones are present and how they change.
TCR/BCR sequencing exists because immune biology is fundamentally organized around clonality, specificity, and expansion.
Standard RNA-seq may tell you that lymphocytes are activated. Repertoire sequencing tells you whether that activation is polyclonal or oligoclonal, stable or shifting over time, and potentially driven by specific antigen-recognizing clones. That makes it a distinct measurement strategy, not just a subtype of RNA sequencing.
Tradeoffs
- Focused on receptor biology rather than broader transcriptome state
- Interpretation can be complex without clinical or antigen context
- Pairing alpha/beta or heavy/light chains may require specialized assay designs
- Not all clonotypes can be linked directly to antigen specificity without further evidence
Bottom line: Immune repertoire sequencing exists because many immunology questions are really about clonality and receptor diversity, not just expression levels.
12) Spatial + Single-Cell Multiome / Multimodal Assays: Why measure more than one molecular layer at once?
What it captures
Single-cell multiome and multimodal assays measure multiple types of biological information from the same cell—or, in spatial multi-omics, from the same tissue context. Depending on the platform, this can include combinations such as:
- RNA + chromatin accessibility,
- RNA + surface proteins,
- RNA + immune receptor sequences,
- RNA + DNA variation,
- or spatially resolved transcriptome + proteome/epigenetic readouts.
Why choose it
You choose multimodal assays when the question is:
- “Is one molecular layer enough to explain this cell state?”
- “Can we connect expression, regulation, and identity directly?”
- “Do we need to know both what a cell is doing and how that state is controlled?”
- “Can we preserve tissue context while measuring more than one type of signal?”
The deeper “why”
A single modality often captures only one slice of biology. RNA can tell you what genes are being expressed, but not always which regulatory elements enabled that state. Chromatin accessibility can suggest regulatory potential, but not always whether the downstream transcript program is being executed. Protein markers can help define phenotype, but may not explain the transcriptional logic behind it.
Multimodal assays exist because many important biological questions are really about relationships between layers, not any one layer alone.
This is especially useful when you want to connect cell identity to regulatory state, transcriptional programs to chromatin control, immune phenotype to clonality, or spatial tissue architecture to molecular function. In practice, these assays help answer not just “what is changing?” but “how is it being controlled, and where is it happening?”
Tradeoffs
- Higher cost and more complex library preparation than single-modality assays
- Lower throughput or shallower depth per modality in some designs
- Stronger computational burden for integration, normalization, and interpretation
- Sample quality requirements can be stricter
- Cross-modality tradeoffs may limit performance compared with assays optimized for one layer alone
Bottom line: Multimodal assays exist because complex biology is rarely fully explained by one molecular layer, and combining signals can reveal mechanisms that single-modality assays miss.
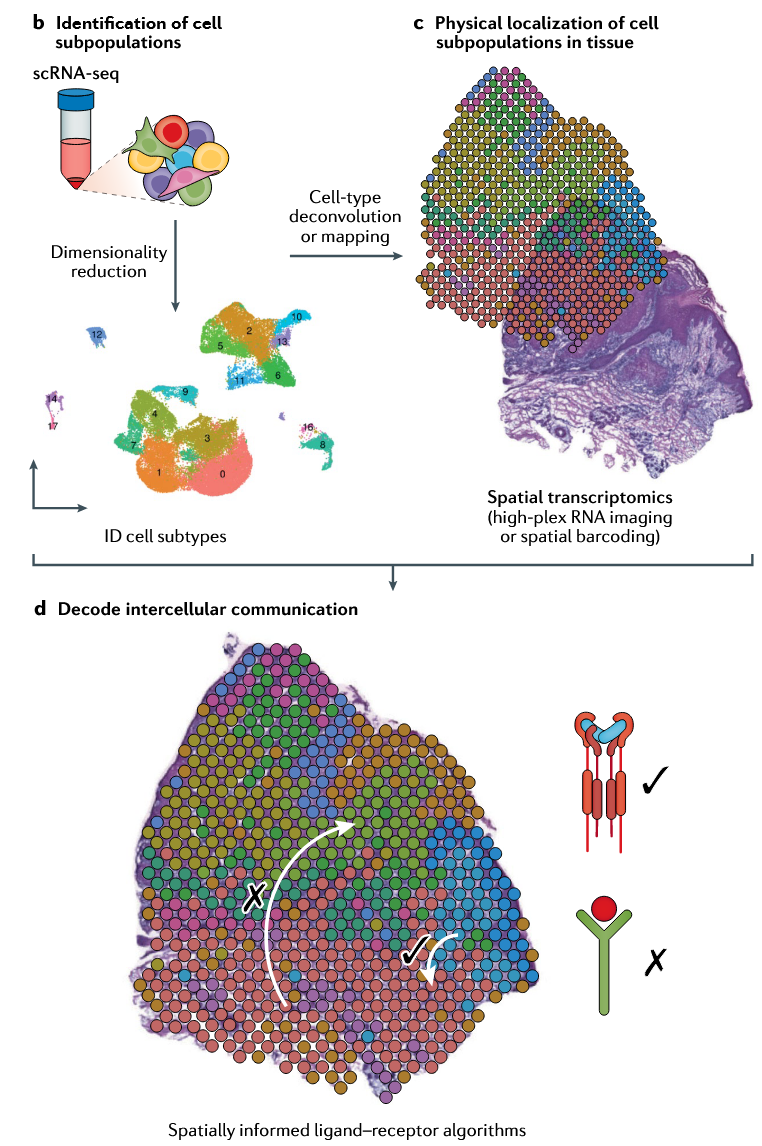
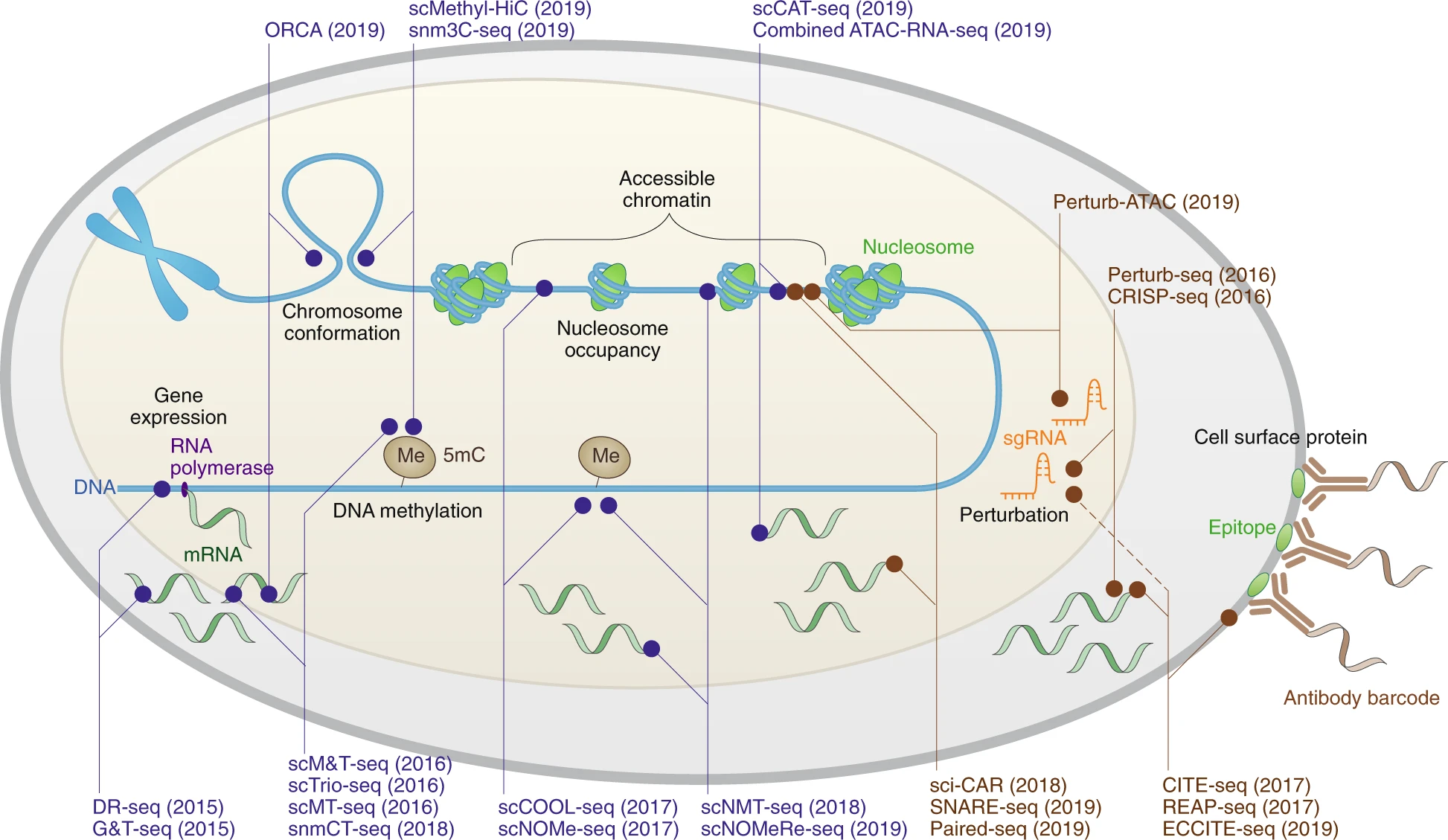
The real framework: sequencing modalities are tradeoff engines
The wrong way to think about sequencing is:
- WGS > WES > panel (as if one simply upgrades the other)
The right way is:
- Each modality optimizes a different axis of truth-finding
For example:
- If you need rare variant sensitivity, you often prioritize targeted depth
- If you need novel discovery, you prioritize breadth
- If you need functional insight, you prioritize RNA/epigenomics
- If you need heterogeneity, you prioritize single-cell
- If you need serial monitoring, you prioritize liquid biopsy-compatible assays
- If you need spatial context, you prioritize spatial transcriptomics
- If you need community composition, you prioritize metagenomic (or amplicon for marker-based profiling)
- If you need clonality and receptor diversity, you prioritize TCR/BCR repertoire
- If you need multiple molecular layers in the same cell, you prioritize multiome/multimodal assays
This is why experienced genomic scientists ask “what decision will this data support?” before they ask “which sequencer will run it?”
Why this matters in real-world genomics teams
In research, clinical genomics, biotech, and diagnostics, sequencing modality selection affects:
- Scientific validity (did we measure the right thing?)
- Clinical utility (can this result change management?)
- Operational efficiency (cost, TAT, scale)
- Regulatory/validation burden
- Bioinformatics complexity
- Interpretation workload
- Product-market fit (for genomics companies)
A team with deep knowledge of NGS and molecular biology doesn’t just know how to run assays. They understand why an assay is the right measurement for a biological mechanism and a downstream decision.
That’s the difference between generating data and generating answers.
Final thought: start with biology, not the instrument
The best sequencing strategy is almost never chosen by brand, trend, or buzzword. It’s chosen by respecting the biology and the question.
Before selecting a modality, ask:
- What decision am I trying to make?
- What biological signal best supports that decision?
- What variant class or molecular layer could invalidate my conclusion if I ignore it?
- What tradeoffs am I willing to accept?
That is the “why” behind sequencing modalities.
And once you start thinking this way, sequencing stops looking like a list of techniques—and starts looking like what it really is: experimental design in service of biological truth.
Comments
Leave a comment using your GitHub account. Your feedback is appreciated!